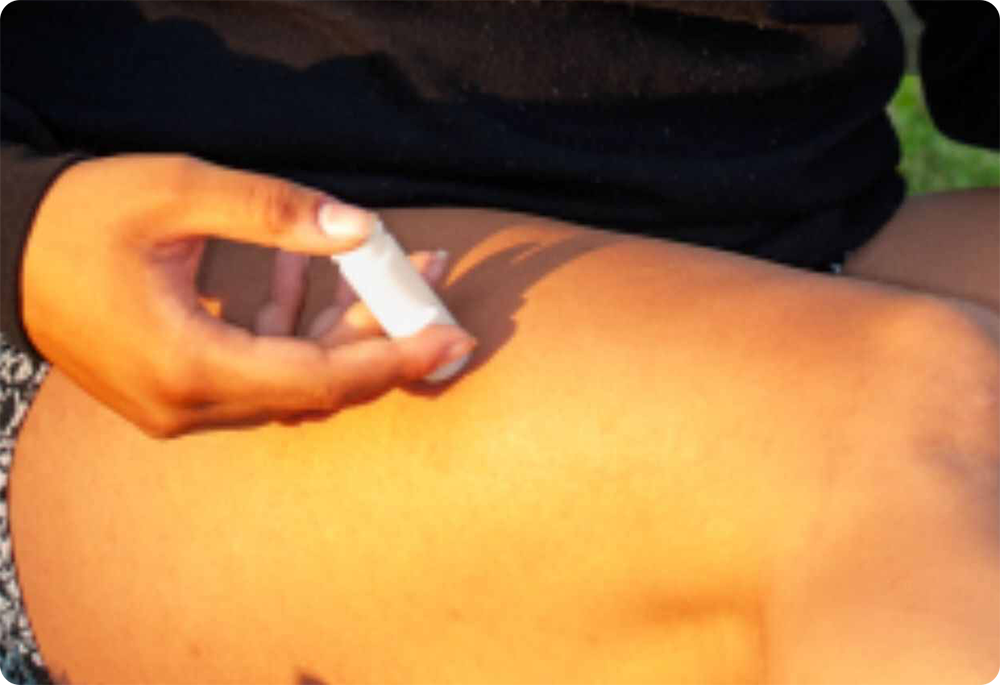
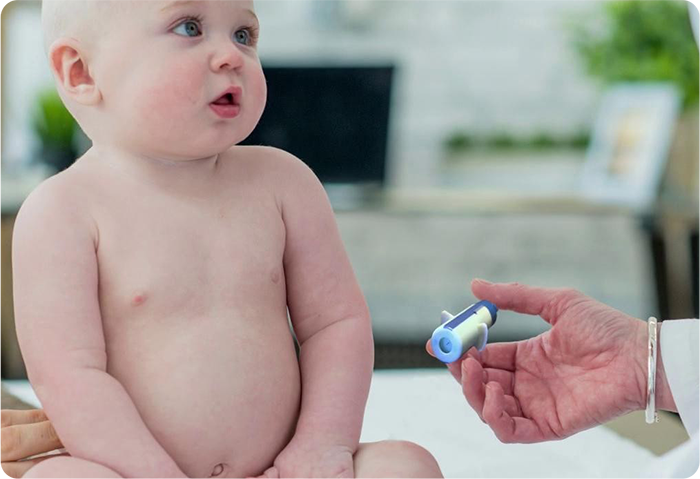
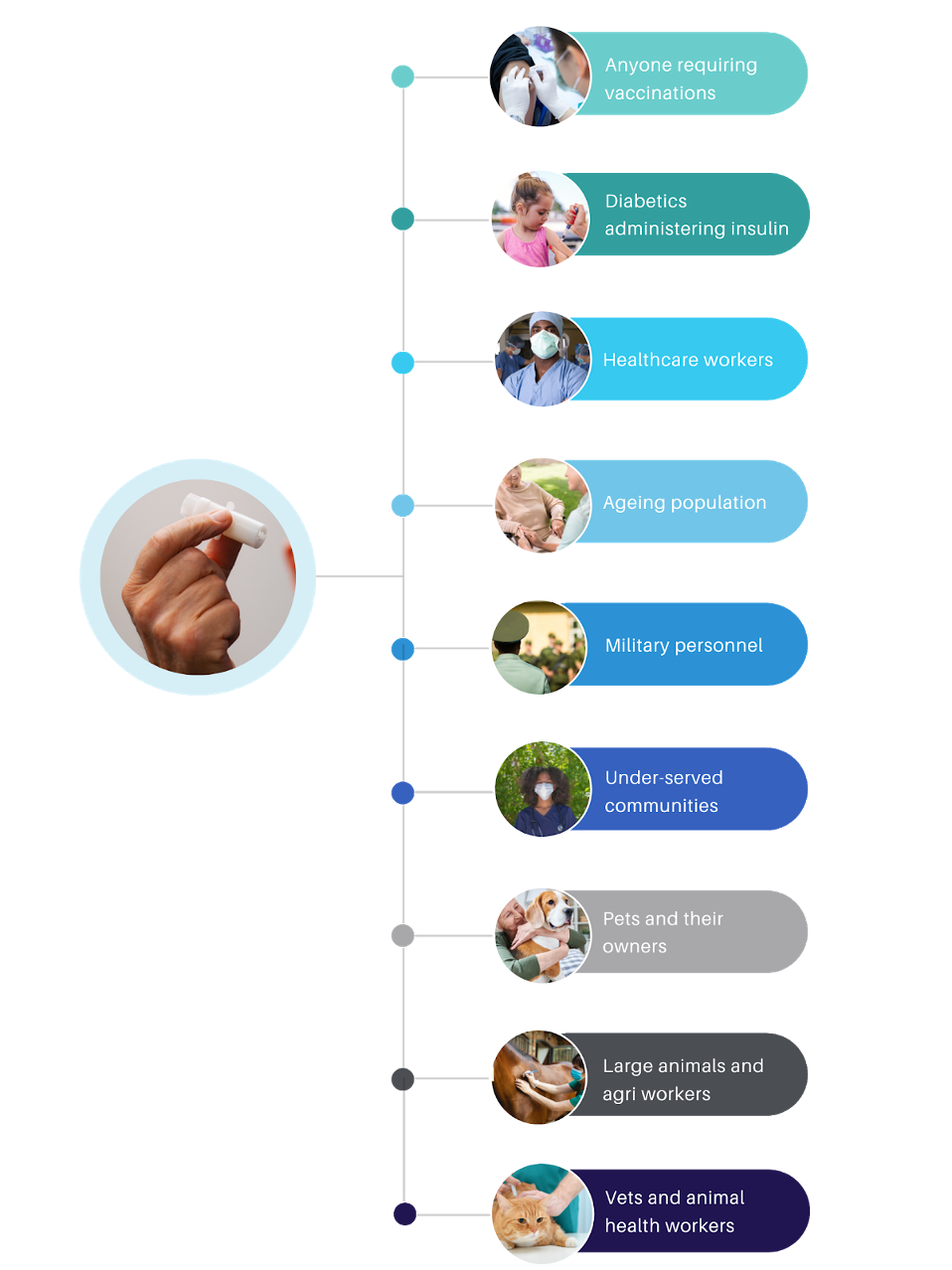
Do you know this is a pain-free injection device for medications and vaccinations?
With more patients around the world requiring injections for a variety of health reasons, the timing has never been better to introduce a safer, more patient-friendly delivery system for important, life-giving medications.